
Stability analysis of ICA components for transcriptomic data¶
Here we propose a short analysis of the stability and the reproductibility of ICA components extracted from several gene expression data sets. We are mainly interested in studying the behavior of transcriptomic data extracted from “Defining the Biological Basis of Radiomic Phenotypes in Lung Cancer” Grossman et al. 2017.
[1]:
#%load_ext autoreload
#%autoreload 2
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import time
0. Load data sets¶
Grossman data set¶
These data sets were extracted from “Defining the Biological Basis of Radiomic Phenotypes in Lung Cancer” Grossman et al. 2017.
df1 contains the expression of 21,766 unique genes for 269 patients with Non-small cell lung cancer (NSCLC) treated at the H. Lee Moffitt Cancer Center, Tampa, Florida, USA. df2 contains the expression of the same 21,766 unique genes for 89 patients with Non-small cell lung cancer (NSCLC)treated at MAASTRO clinical, Maastricht, NL. Gene expression values were measured on a custom Rosetta/Merck Affymetrix 2.0 microarray chipset and normalized with the robust multi-array average (RMA) algorithm.
[2]:
df1 = pd.read_excel("data/data_USA.xlsx" , sheet_name = 'expression' , index_col= 0).transpose()
df2 = pd.read_excel("data/data_UE.xlsx" , sheet_name = 'expression' , index_col= 0).transpose()
#With xlrd >= 2.0.0 you will need to use a different engine since xrld removed support for anything
#other than .xls files (make sure openpyxl is installed). It will usually take a bit of time.
#df1 = pd.read_excel("data/data_USA.xlsx" , engine = 'openpyxl' , sheet_name = 'expression' , index_col= 0).transpose()
#df2 = pd.read_excel("data/data_UE.xlsx" , engine = 'openpyxl' , sheet_name = 'expression' , index_col= 0).transpose()
dic = {}
for col in df1.columns:
dic[col] = int(col.split('.')[1])
df1 = df1.rename(columns = dic)
df1['Dataset'] = ['Grossman USA']*df1.shape[0]
df1 = df1.reset_index().rename(columns = {'index' : 'Samples'}).set_index(['Dataset' , 'Samples'])
df2 = df2.rename(columns = dic)
df2['Dataset'] = ['Grossman UE']*df2.shape[0]
df2 = df2.reset_index().rename(columns = {'index' : 'Samples'}).set_index(['Dataset' , 'Samples'])
df1.head()
[2]:
| 3643 | 84263 | 7171 | 2934 | 11052 | 1241 | 6453 | 57541 | 9349 | 11165 | ... | 643669 | 1572 | 8551 | 26784 | 26783 | 26782 | 26779 | 26778 | 26777 | 100132941 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | Samples | |||||||||||||||||||||
| Grossman USA | RadioGenomic-017 | 5.205151 | 7.097989 | 9.559617 | 8.396808 | 7.603719 | 7.990605 | 10.044401 | 9.054930 | 7.383169 | 8.177010 | ... | 6.419273 | 3.809826 | 6.507880 | 6.572121 | 5.400848 | 5.951391 | 3.381860 | 9.825584 | 2.905091 | 5.622438 |
| RadioGenomic-055 | 5.615738 | 6.585052 | 9.777869 | 9.082415 | 8.639498 | 6.781274 | 9.541826 | 8.866110 | 6.422702 | 7.196294 | ... | 5.753828 | 4.186127 | 6.821582 | 7.031406 | 4.852417 | 6.140850 | 2.629760 | 9.005145 | 3.366466 | 5.495330 | |
| RadioGenomic-227 | 5.679276 | 7.747854 | 10.648704 | 9.127985 | 7.369421 | 7.203773 | 8.972255 | 8.328371 | 7.269232 | 7.449183 | ... | 5.666999 | 4.316130 | 6.637855 | 6.248824 | 4.664228 | 5.767970 | 2.911470 | 8.674466 | 3.337194 | 6.308605 | |
| RadioGenomic-222 | 5.317341 | 7.196276 | 10.949771 | 8.098896 | 7.639882 | 7.971876 | 10.159637 | 8.667702 | 8.474250 | 7.271477 | ... | 5.531060 | 3.403776 | 7.059419 | 6.201873 | 4.690005 | 6.256286 | 4.119688 | 9.099659 | 3.181781 | 5.740033 | |
| RadioGenomic-212 | 7.196904 | 9.346492 | 9.673778 | 9.358636 | 8.741693 | 7.616498 | 10.376653 | 8.701461 | 6.601991 | 7.344651 | ... | 5.519642 | 3.796049 | 7.332635 | 6.050121 | 4.898523 | 6.537895 | 3.600895 | 8.792510 | 2.945391 | 5.835411 |
5 rows × 21766 columns
Lim NSCL data set¶
Note : In order to be able to join this data set with the Grossman data set, we converted the official gene symbols to entrez ids. We used the Database for Annotation, Visualization and Integrated Discovery(DAVID)
[3]:
new_data = pd.read_excel("data/new_data.xlsx" , sheet_name=['expression' , 'clinical'] , index_col=0)
#With xlrd >= 2.0.0 you will need to use a different engine since xrld removed support for anything
#other than .xls files (make sure openpyxl is installed). It will usually take a bit of time.
#new_data = pd.read_excel("data/new_data.xlsx" , engine = 'openpyxl' , sheet_name=['expression' , 'clinical'] , index_col=0)
new_data['expression'] = new_data['expression'].drop(columns = 'gene_name').transpose()
temp = new_data['expression'].join(new_data['clinical'][['Dataset' , 'Histology (ADC: adenocarcinoma; LCC: large cell carcinoma; SCC: squamous cell carcinoma)']])
df3 = temp.reset_index().rename(columns = {'index' : 'Samples'}).set_index(['Dataset' , 'Samples'])
del temp , new_data
df3 = df3[df3['Histology (ADC: adenocarcinoma; LCC: large cell carcinoma; SCC: squamous cell carcinoma)'] != 'Healthy'].drop(columns = 'Histology (ADC: adenocarcinoma; LCC: large cell carcinoma; SCC: squamous cell carcinoma)')
df3.head()
[3]:
| 1 | 2 | 144571 | 65985 | 13 | 201651 | 51166 | 195827 | 79719 | 22848 | ... | 9183 | 55055 | 11130 | 7789 | 158586 | 440590 | 79699 | 7791 | 23140 | 26009 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | Samples | |||||||||||||||||||||
| GSE33356 | GSM494556 | 5.234802 | 11.922797 | 5.031032 | 9.141089 | 9.934807 | 6.868094 | 5.038721 | 6.845863 | 7.877033 | 9.201101 | ... | 7.149422 | 7.737312 | 8.386004 | 5.994052 | 7.685643 | 4.546110 | 8.870484 | 7.938569 | 7.596789 | 8.049103 |
| GSM494557 | 5.069035 | 11.146006 | 4.737330 | 8.926558 | 4.419542 | 3.276470 | 7.517926 | 6.945466 | 7.387878 | 9.279903 | ... | 7.307666 | 7.693829 | 9.986055 | 5.808656 | 5.968874 | 4.979624 | 9.177783 | 7.976926 | 7.044507 | 8.192161 | |
| GSM494558 | 5.514972 | 12.378934 | 6.990395 | 7.268946 | 4.253987 | 2.978153 | 6.286073 | 7.201963 | 7.125318 | 10.170862 | ... | 6.886556 | 7.927103 | 8.124558 | 5.526909 | 6.000892 | 4.951939 | 8.613027 | 8.642108 | 7.747791 | 7.947040 | |
| GSM494559 | 6.871695 | 11.853862 | 4.589314 | 8.874169 | 5.911853 | 4.412516 | 6.304975 | 7.731857 | 7.300380 | 8.649045 | ... | 7.939107 | 8.589020 | 9.673297 | 6.190925 | 6.736635 | 4.765782 | 9.643763 | 7.295604 | 7.839339 | 8.377581 | |
| GSM494560 | 6.761781 | 11.200515 | 5.001184 | 9.027746 | 5.356248 | 3.515208 | 5.545497 | 7.552539 | 7.309306 | 9.229319 | ... | 7.833734 | 8.689572 | 10.304345 | 6.090843 | 6.703918 | 5.821131 | 9.187349 | 7.557380 | 7.320086 | 8.827688 |
5 rows × 10077 columns
We only keep data sets with more than 89 samples to avoid ending up with very noisy ICA components which will compromise the stability analysis.
[4]:
dataframe = pd.concat([df1, df2 , df3], join = 'inner')
names = list(dataframe.index.unique(0))
exclude = []
print("Data sets in the study : ")
for name in names:
n_samples = dataframe.loc[name].shape[0]
if n_samples < 89:
exclude.append(name)
else:
print(name , n_samples)
del df1 , df2 , df3
names = list(set(names) - set(exclude))
Data sets in the study :
Grossman USA 262
Grossman UE 89
GSE19188 91
GSE28571 100
GSE50081 181
GSE31210 226
GSE18842 91
1. Most Stable Transcriptome Dimension (MSTD)¶
In order to select the number of ICA components we refer to “Determining the optimal number of independent components for reproducible transcriptomic data analysis” Kairov et al. 2017. The idea is to find a trade-off between a sufficiently large number of components to capture all essential information and a restricted dimension to avoid ending up with a lot of very unstable components which only capture noise.
To do so we plot the stability distribution for a wide range of different number of components M = 5, … , 100 and we observe which choice of M most satisfies the trade-off the most.
[5]:
#MSTD plot for the data set GSE31210.
from sica.base import MSTD
X = dataframe.loc['GSE31210'].apply(func = lambda x : x - x.mean() , axis = 0)
MSTD(X , 5 , 100 , 2 , 20 , max_iter = 3000)
#MSTD(X.values , 5 , 100 , 2 , 20 , max_iter = 3000)
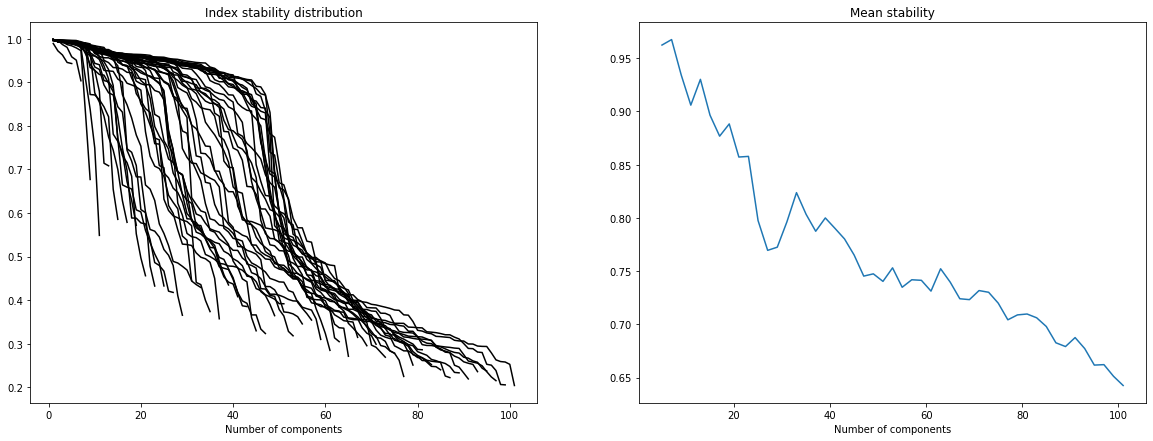
[5]:
MSTD = 45
2. Stabilized ICA decompostion for each data set¶
Our stabilized ICA algorithm is a copy of the ICASSO algorithm (implemented in MATLAB) “ICASSO: software for investigating the reliability of ICA estimates by clustering and visualization” Himberg et al. The main idea is to iterate several times the FastICA algorithm (ex: from sklearn), cluster the results (agglomerative hierarchical clustering) and define the final ICA components as the centrotype of each cluster.
[6]:
from sica.base import StabilizedICA as sICA
[7]:
Sources = []
decomp = sICA(n_components = MSTD , n_runs = 30, n_jobs = -1)
for name in names:
start = time.time()
X = dataframe.loc[name].apply(func = lambda x : x - x.mean() , axis = 0)
#decomp.fit(X.values)
decomp.fit(X)
Sources.append(decomp.S_ )
end = time.time()
minutes, seconds = divmod(end - start, 60)
print(name + " done !" , "running time (min): " + "{:0>2}:{:05.2f}".format(int(minutes),seconds))
GSE31210 done ! running time (min): 00:41.43
GSE50081 done ! running time (min): 00:27.18
Grossman USA done ! running time (min): 00:32.93
GSE19188 done ! running time (min): 00:25.28
GSE28571 done ! running time (min): 00:23.74
GSE18842 done ! running time (min): 00:21.22
Grossman UE done ! running time (min): 00:24.44
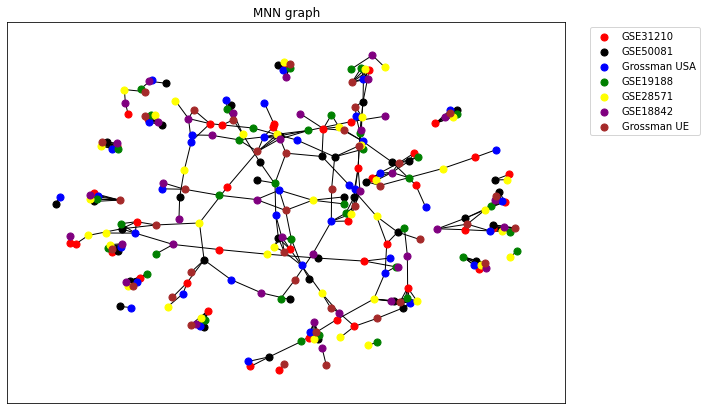
3. Study of reproducibility - MNN graph¶
In order to study the reproducibility of ICA components between the four data sets we use the notion of Reciprocal Best Hit (RBH). We consider that a component \(i\) extracted from a data set D1 and another component \(j\) extracted from a data set D2 are linked if:
\begin{equation} |\rho_{ij}| = \max \{ |\rho_{ik}| , \forall k \, \in D2 \} = \max \{ |\rho_{lj}| , \forall l \, \in D1 \} \end{equation}
Here \(\rho_{ij}\) represents the Pearson’s correlation coefficient between the components \(i\) and \(j\). We chose to use Pearson’s coefficient over non-parametric coefficients such as Kendall’s or Spearman’s coefficients since it is strongly influenced by extreme values. Yet in our case these extreme values characterize our ICA components .
[8]:
from sica.mutualknn import MNNgraph
Note : The layout of the following RBH network is strongly influenced by a random initialisation (Fruchterman-Reingold force-directed algorithm). One may have to run the following result several times to obtain a satisfying result.
[12]:
fig , ax = plt.subplots(figsize = (10 , 7))
cg = MNNgraph(data = Sources , names = names , k=1, weighted=False)
cg.draw(ax=ax, colors = ['r' , 'k' , 'b' , 'g' , 'yellow' , 'purple' , 'brown'] , spacing = 2)
ax.legend(bbox_to_anchor=(1.25 , 1))
[12]:
<matplotlib.legend.Legend at 0x218dbb6deb0>
[14]:
cg.export_json("stability.json")
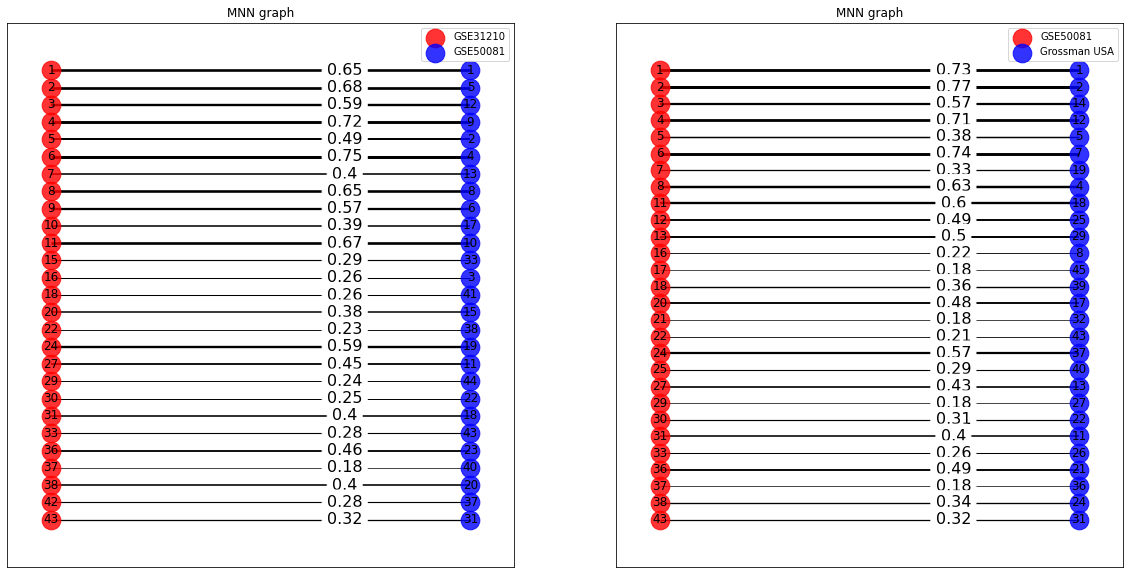
We can also draw the bipartite RBH graph for the ICA components of two data sets. We display the Pearson correlation coefficient for each link. In the following we display the bipartite graphs for the couples of sets (‘Grossman UE’ , ‘GSE19188’) and (‘Grossman UE’ , ‘GSE50081’).
[10]:
fig , axes = plt.subplots(1 , 2 , figsize=(20 , 10))
for i in range(2):
cg = MNNgraph(data = [Sources[0] , Sources[i+1]] , names = [names[i] , names[i+1]] , k=1)
cg.draw(ax=axes[i] , bipartite_graph = True)
4. Cytoscape visualization of the stability network¶
[19]:
import ipycytoscape
import json
with open("stability.json") as fi:
json_file = json.load(fi)
cytoscapeobj = ipycytoscape.CytoscapeWidget()
cytoscapeobj.graph.add_graph_from_json(json_file['elements'])
cytoscapeobj.set_layout(name = 'cose')
[20]:
cytoscapeobj.set_style([{'selector': 'node',
'css': {
'content': 'data(name)',
'font-size': '12px',
'text-valign': 'center',
'text-halign': 'center',
'background-color': 'blue',
'color': 'b',
}
},
{'selector': 'edge',
'css': {
'curve-style': 'haystack',
'haystack-radius': '0.5',
'opacity': '0.7',
'line-color': '#bbb',
'width': 'mapData(weight , 0 , 1 , 1 , 15)',
'overlay-padding': '3px'
}
}])
[21]:
cytoscapeobj